Hepatic encephalopathy: Suspect it early in patients with cirrhosis





ABSTRACTAs viral hepatitis and nonalcoholic fatty liver disease continue to increase in prevalence, we will see more cases of hepatic encephalopathy. Primary care physicians are often the first to suspect it, as they are familiar with the patient’s usual mental and physical status. This serious complication typically occurs in patients with severe comorbidities and requires multidisciplinary evaluation and care.
KEY POINTS
- Hepatic encephalopathy should be considered in any patient with cirrhosis who presents with neuropsychiatric manifestations in the absence of another brain disorder, such as stroke or brain tumor.
- “Minimal” hepatic encephalopathy may not be obvious on clinical examination but can be detected with neurophysiologic and neuropsychiatric testing.
- Every cirrhotic patient is at risk; potential precipitating factors should be addressed during regular clinic visits.
- Management requires prompt identification of precipitating factors and initiation of empiric medical therapy. Current treatments include drugs to prevent ammonia generation in the colon.
- Long-acting benzodiazepines should not be used to treat sleep disorders in patients with cirrhosis, as they may precipitate encephalopathy.
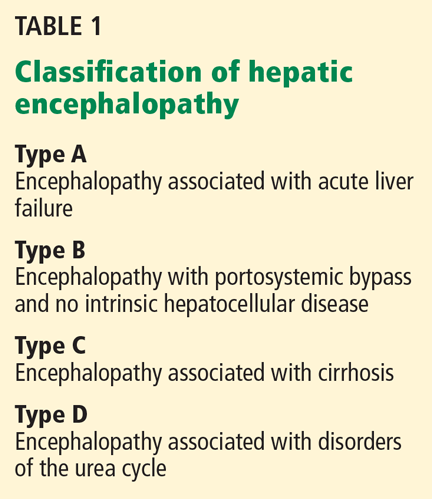
Hepatic encephalopathy is a serious but often reversible complication that arises when the liver cannot detoxify the portal venous blood (Table 1).1
PROPOSED PATHOGENETIC FACTORS
About 5.5 million cases of chronic liver disease and cirrhosis were reported in the United States in 2001. Hepatic encephalopathy is becoming more common as the prevalence of cirrhosis increases,2 and this will have important economic repercussions; in 2001, charges from hospitalizations because of hepatic encephalopathy were estimated at $932 million.3
Hepatic encephalopathy develops as cirrhosis progresses or as a result of portosystemic shunting, so that the liver cannot detoxify the portal venous blood. Several neurotoxins (notably ammonia) and inflammatory mediators play key roles in its pathogenesis, inducing low-grade brain edema and producing a wide spectrum of neuropsychiatric manifestations.4 Yet its pathogenesis is not entirely understood, impeding advances in its diagnosis and therapy.
Several hypotheses about the pathogenesis of hepatic encephalopathy have emerged in the last few years, and a number of factors are reported to directly or indirectly affect brain function in this condition. Ammonia and glutamine are the neurotoxins most often implicated in this syndrome5; others include inflammatory mediators, certain amino acids, and manganese.5,6
Ammonia causes brain swelling
Ammonia is primarily the byproduct of bacterial metabolism of protein and nitrogenous compounds in the colon and of glutamine metabolism in enterocytes.7
Normally, gut-absorbed ammonia is delivered via the portal vein to the liver, where most of it is metabolized into urea, leaving a small amount to be metabolized in the muscles, heart, brain, and kidneys. In cirrhosis and other conditions associated with hepatic encephalopathy, less ammonia is metabolized into urea and more of it reaches the astrocytes in the brain. The brain lacks a urea cycle but metabolizes ammonia to glutamine via glutamine synthase, an enzyme unique to astrocytes.
Ammonia causes swelling of astrocytes and brain edema via generation of glutamine, an osmotically active substance.
Glutamine causes swelling, oxidative stress
Glutamine draws water into astrocytes and induces changes of type II astrocytosis (also called Alzheimer type II astrocytosis)5 characterized by swelling, enlarged and pale nuclei, and displacement of chromatin to the periphery of the cell. Inhibition of glutamine synthase prevents astrocyte swelling in animals.8
Glutamine also enhances the activation of several receptors, including N-methyl-d-aspartate (NMDA) receptors,9,10 gammaaminobutyric acid (GABA) receptors, and peripheral-type benzodiazepine receptors on the mitochondrial membrane.10–12 A state of oxidative stress ensues, and this affects oxidation of protein and RNA, neurotransmitter synthesis, and neurotransmission at the neuronal junction.13 Reactive nitrogen and oxide radicals induce the release of inflammatory mediators such as interleukins 1 and 6, tumor necrosis factor, interferons, and neurosteroids, and contribute to edema and neurotoxicity.6,10 Neurosteroids are byproducts of mitochondrial metabolism of steroid hormones in the astrocyte.
Manganese enhances neurosteroid synthesis
Manganese enhances neurosteroid synthesis via activation of translocator proteins on the astrocyte membrane. It was first recognized as a factor in hepatic encephalopathy when cirrhotic patients experiencing extrapyramidal symptoms were found to have deposits of manganese in the caudate nucleus and in the globus pallidus on magnetic resonance imaging (MRI). Such deposits were also seen in specimens of brain tissue on autopsy of these patients. When the encephalopathy resolved, so did the abnormalities on MRI.14,15
Changes in the blood-brain barrier
Astrocytes contribute to the selective permeability of the blood-brain barrier. Disruptions in the permeability of the blood-brain barrier underlie hepatic encephalopathy, with poor diffusion of molecules out of astrocytes.
For instance, zinc, which plays a regulatory role in gene transcription and synaptic plasticity, accumulates in the astrocytes, causing relative zinc deficiency and further affecting neurotransmitter synthesis and neurotransmission at the neuronal synapse.6,16