Pink Papulonodular Eruption on the Trunk and Arms





THE DIAGNOSIS: Sarcoidlike Reaction
Sarcoidlike reaction (SLR) is a rare cutaneous immune-related adverse event characterized by a multisystem granulomatous reaction indistinguishable from sarcoidosis but temporally associated with a trigger.1 Drug-induced SLR typically involves the mediastinal or hilar lymph nodes, with frequent involvement of the lungs and skin; cutaneous manifestations typically encompass erythematous papulonodular eruptions on the trunk and extremities.1-3 Sarcoidosis predominantly affects middle-aged women of African American or Scandinavian descent; genetic predisposition likely is a contributing factor.4 Unlike sarcoidosis, SLR is linked to various triggers such as medication or malignancy.
Immune checkpoint inhibitors (ICIs), particularly anti–PD-1 agents, have been linked to SLR through overexpression of proinflammatory cytokines, resulting in excessive T-helper 1 cell and macrophage activation and granulomatous eruption; notably, cutaneous immune-related adverse events often are correlated with greater treatment efficacy.5,6 Overall, anticancer therapy–induced SLR is most commonly reported in patients receiving ICIs for melanoma but it also has been described with ICI therapy for other cancers and with chemotherapy for melanoma. 1,3 Although most cases demonstrate both cutaneous and extracutaneous involvement, approximately 13 reported cases have been exclusively cutaneous.1 Recognition of SLR is important because misdiagnosis as true sarcoidosis may prompt unnecessary testing or therapy; furthermore, distinction from tumor progression is critical.3 The lesions can mimic other granulomatous or inflammatory dermatoses, posing a diagnostic challenge.
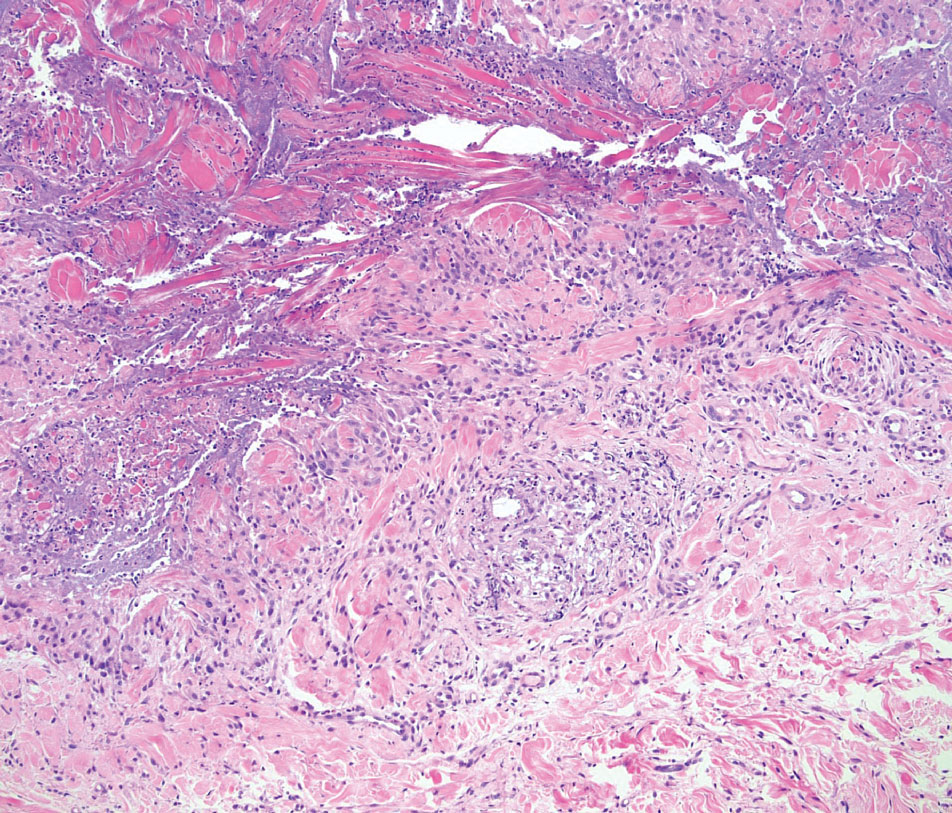
On histopathology, SLR typically demonstrates well-formed, noncaseating dermal granulomas composed of epithelioid histiocytes and Langhans or foreign-body giant cells, a sparse lymphocytic rim, and few plasma cells.2,4 Immunohistochemistry shows CD68-positive histiocytes predominating within the granulomas. Asteroid and Schaumann bodies occasionally are present.7 Special stains will be negative for microorganisms. Sarcoidosis manifests essentially identically from both a clinical and histopathologic perspective (Figure 1). Temporal association with an offending agent and symptomatic resolution following drug cessation remain the most reliable features for distinguishing SLR from sarcoidosis.7
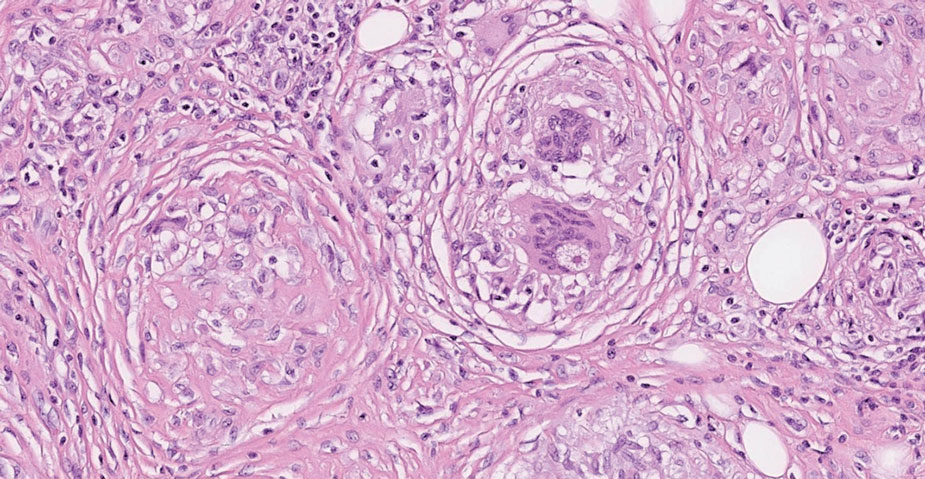
Tuberculoid leprosy is a chronic infectious disease caused by Mycobacterium leprae (found most commonly in tropical regions) and manifesting as localized hypopigmented macules or papules with raised erythematous margins.8 Histopathologically, lesions show well-formed granulomas composed of epithelioid histiocytes and Langhans giant cells without necrosis, surrounded by a prominent lymphocytic rim (Figure 2).9 Rarely, focal caseous necrosis occurs, particularly in involved nerves.10 Hallmark features include enlarged cutaneous nerves surrounded by dermal granulomas and absence of bacilli on special stains; eccrine glands are infrequently involved.9 Standard treatment is 6 months of combination therapy with dapsone and rifampin.
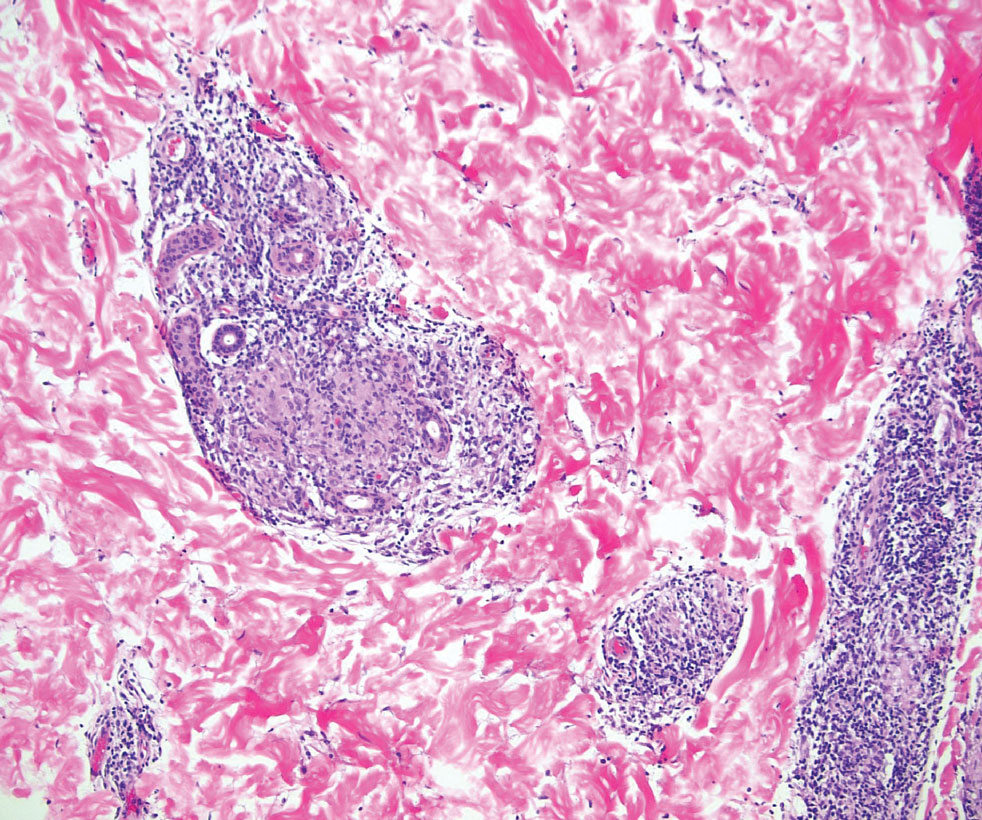
Generalized granuloma annulare is an inflammatory dermatosis manifesting as diffuse erythematous annular papules, classically on the trunk and extremities.11 It predominantly affects individuals in their fifth and sixth decades of life and may be drug induced.2 Histopathology may reveal palisaded granulomas with central necrobiotic collagen, intercalating histiocytes, and interstitial mucin (Figure 3).2 Pathology also may show interstitial histiocytes and lymphocytes intercalating between collagen bundles with increased mucin but absent palisading or necrobiosis or a mixed pattern.2,12 Alcian blue or colloidal iron stains highlight mucin to help distinguish from other granulomatous processes. Multinucleated giant cells are rare. The nonnecrobiotic histologic pattern can mimic sarcoidosis, necessitating clinical correlation for correct diagnosis.13 Certain cases show genetic predisposition, such as HLA-B35, with a relapsing course often requiring combined systemic immunosuppression and phototherapy.14
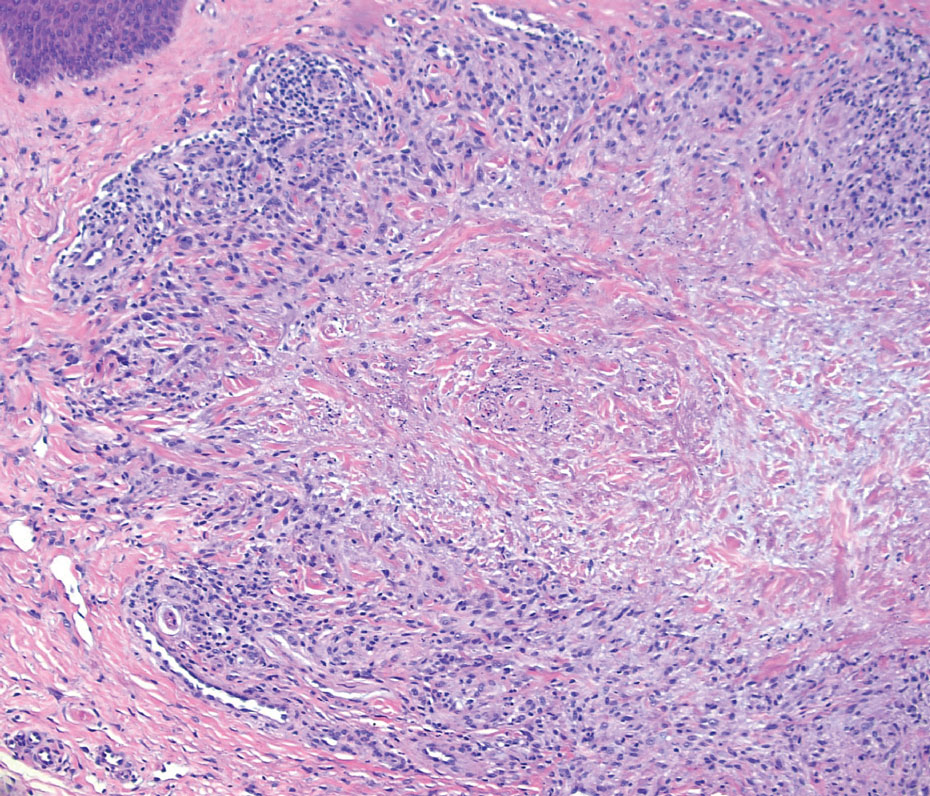
Granulomatosis with polyangiitis is a systemic vasculitis that classically manifests as palpable purpura on the lower extremities, often with ulceration. Localized erythematous papules on the extensor surfaces may occur less commonly.15 Pathogenesis involves antineutrophil cytoplasmic antibodies inducing neutrophil degranulation, release of reactive oxygen species and proinflammatory cytokines, and subsequent endothelial damage.15 Histopathology shows necrotizing granulomatous inflammation and necrotizing vasculitis of small and medium vessels with nuclear debris.15 Poorly formed granulomas containing abundant neutrophils and mixed perivascular inflammatory infiltrates may be seen with or without vasculitis (Figure 4). Systemic features commonly include chronic rhinosinusitis, pauci-immune glomerulonephritis, and pulmonary nodules.15 Pharmacotherapy includes glucocorticoids combined with a glucocorticoid-sparing agent.