Cordlike Dermal Plaques and Nodules on the Neck and Hands





The Diagnosis: Fibroblastic Rheumatism
Routine histologic sections stained with hematoxylin and eosin demonstrated a noncircumscribed dermal proliferation of fibroblasts and myofibroblasts with thickened collagen bundles (Figure, A and B). Focally fragmented elastin fibers were noted with Verhoeff elastic tissue stain. Alcian blue stain did not show increased dermal mucin. With the clinical presentation and histologic findings described, we diagnosed the patient with fibroblastic rheumatism (FR). To date, the patient's condition has stabilized overall with skin lesions fading and minimal to no joint pain. Current therapies include adalimumab, mycophenolate mofetil 500 mg 3 times daily, and low-dose prednisone.
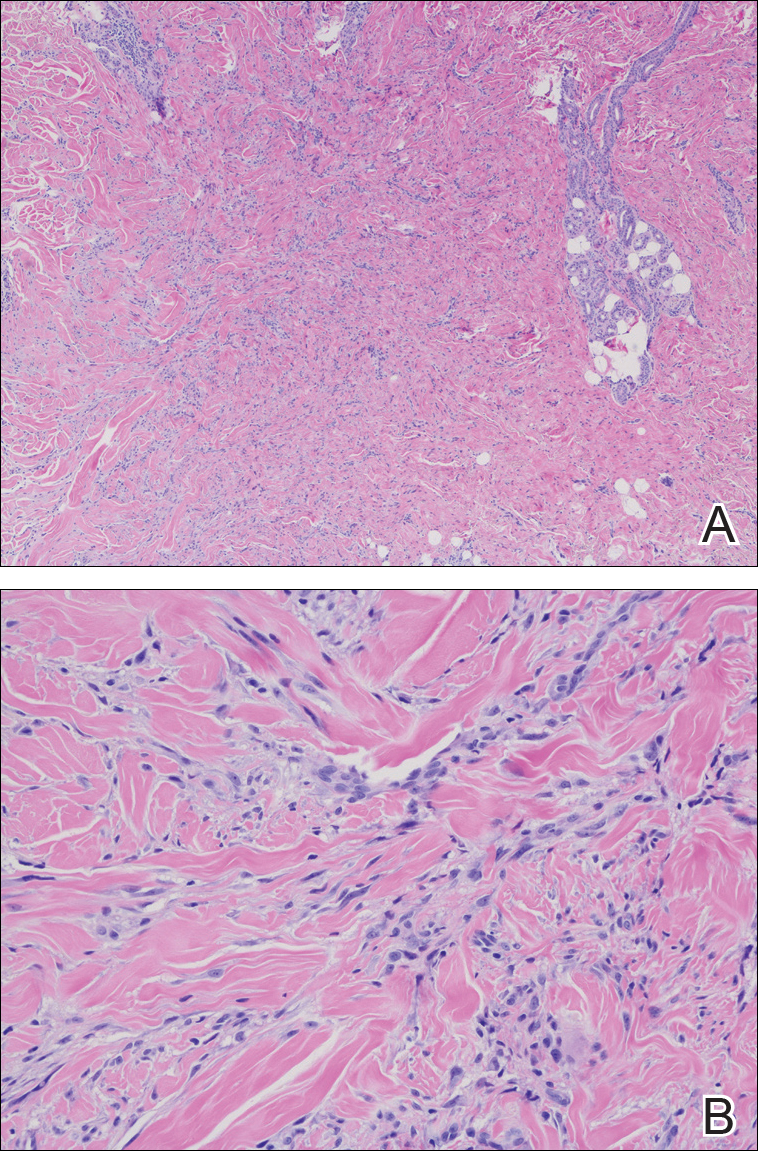
Fibroblastic rheumatism is a rare arthropathy with cutaneous findings initially described by Chaouat et al1 in 1980. Age of onset varies, and the condition also has been observed in pediatric patients.2 Fibroblastic rheumatism is characterized by sudden onset of firm, flesh-colored, subcutaneous nodules on periungual and periarticular surfaces.2 Neck lesions rarely are described,2-4 and cordlike plaques previously have not been reported in FR. Typically, patients develop diffusely swollen fingers, palmar thickening, sclerodactyly, and contractures. The eruption may be accompanied by Raynaud phenomenon as well as a progressive symmetric erosive arthropathy.2,5
The clinical course in FR is variable. The cutaneous findings spontaneously may regress in months to years.3,4 However, polyarthropathy often is destructive and progresses to disability.3 Response to therapy has been unpredictable, and the following treatments have been tried, generally with poor efficacy: aspirin, nonsteroidal anti-inflammatory drugs, hydroxychloroquine, colchicine, methotrexate, prednisone, infliximab, D-penicillamine, interferon alfa, and intensive physical therapy.2-4,6 Histologic characteristics may include thickened collagen bundles along with a fibroblastic and myofibroblastic proliferation. Elastic fibers may be decreased or absent.2,3,5
,Clinical and histologic features in FR may mimic other entities; thus, clinical pathological correlation is essential in determining the correct diagnosis. Considerations in the differential diagnoses include multicentric reticulohistiocytosis (MRH), palisaded neutrophilic and granulomatous dermatitis, and scleroderma.
In MRH, a symmetric erosive arthritis of mainly distal interphalangeal joints typically precedes the cutaneous disease. Occurrence of arthritis mutilans is reported in approximately half of patients.4 Cutaneous manifestations typically include the presence of coral bead-like papules and nodules over the dorsal aspect of the hands, face, and neck. Unlike FR, MRH has a concomitant autoimmune disease in up to 20% of cases and an associated malignancy in up to 31% of cases, with breast and ovarian carcinomas most common. On histology, MRH is characterized by a nodular infiltrate of histiocytes and multinucleated giant cells with eosinophilic ground-glass cytoplasm.4 No notable collagen changes or fibroblastic proliferations typically are present.
Palisaded neutrophilic and granulomatous dermatitis, usually associated with rheumatoid arthritis or connective tissue disease, classically presents as annular plaques and indurated linear bands over the trunk and extremities. However, its clinical presentation is quite variable and may include pink to violaceous urticarialike; livedoid-appearing; or nonspecific papules, plaques, or nodules. Histology in palisaded neutrophilic and granulomatous dermatitis shows a dense dermal neutrophilic infiltrate associated with interstitial histiocytes having a palisading arrangement around degenerated collagen.7 No fibroblastic proliferation typically is present.
Scleroderma can be distinguished based on additional clinical and laboratory findings as well as histology showing thickened collagen bundles without fibroblastic proliferation.2 The histologic findings also may suggest inclusion of dermatofibroma or a scar in the differential diagnosis, though the clinical presentation of these entities would not support these diagnoses.
Acknowledgments
We thank the patient for granting permission to share this information. We also thank Sheng Chen, MD, PhD (Lake Success, New York), for his dermatopathological contributions to the case.